
Welcome to emmahodcroft.com
I am a post-doctoral researcher at the ISPM, University of Bern in Switzerland, working with Christian Althaus. The majority of my past research has been on the phylogenetics, molecular epidemiology, and simulation of HIV, and I was previously involved in the PANGEA_HIV initiative, funded by the Bill & Melinda Gates Foundation.
I'm now working on nextstrain. I previously was deeply involved in expanding Nextstrain to work with bacteria (doing some tuberculosis work in the process), and was part of a major 'modular' refactor of the 'augur' and 'auspice' code. More recently I've been working on Enterovirus-D68 - check out our Nextstrain.org build here.
At the moment, I am working full-time on SARS-CoV-2 in a number of different ways, but am proud to be part of the Nextstrain team
supporting the SARS-CoV-2 builds.
(More information in 'Research' below.)
The most exciting phrase to hear in science, the one that heralds
the most discoveries, is not "Eureka!" but "That's funny..."
Current

I'm a co-developer as part of the nextstrain team, headed up by Richard Neher and
Trevor Bedford, where I have
been working since November 2017 on 'the next step for Nextstrain'. I have mostly focused on Enterovirus D68, a respiratory pathogen
that primarily infects young children. In recent years, it has caused more severe respiratory infections and sometimes paralysis, and seems to circulate in
in the US and Europe in the autumns of even-numbered years.
You can check out our recent pre-print of our latest findings here,
and view our live-build of EV-D68 here.
My previous work on this was expanding it to bacterial pathogens. Some of the challenges
I've tackled include finding computationally and memory efficient ways to handle the much larger genomes, working with much slower
mutation rates, detecting drug resistance, and handling plasmids and horizontal gene-transfer.
Tuberculosis, with its lack of plasmids and recombination, huge public health impact, and rise in resistance, is serving as the 'pilot'
organism for this endevor, though we have run other bacterial pathogens, like Campylobacter.
Previous
 The majority of my previous work has focused on HIV. I was part of the PANGEA_HIV
initiative, funded by the Bill & Melinda Gates Foundation. PANGEA_HIV aimed to use
phylogenetics and molecular epidemiology to gain new understanding of the HIV epidemic
in sub-Saharan Africa.
The majority of my previous work has focused on HIV. I was part of the PANGEA_HIV
initiative, funded by the Bill & Melinda Gates Foundation. PANGEA_HIV aimed to use
phylogenetics and molecular epidemiology to gain new understanding of the HIV epidemic
in sub-Saharan Africa. I created a stochastic, agent-based model (DSPS-HIV) that simulates HIV epidemics, which I used to generate data sets that can be used to assess phylogenetic methods. Disease stage and transmission risk are dependant on viral load, and contact networks are highly customizable.
You can see the simulated datasets released as part of PANGEA_HIV work package 4 here. You can read more details about the DSPS-HIV, as well as a full description of the PANGEA_HIV methods comparison exercise in our recent publication in MBE.
"But all evolutionary biologists know that variation itself is nature's only irreducible essence. Variation is the hard reality, not a set of imperfect measures for a central tendency. Means and medians are the abstractions."
Post-Graduate
-
I completed my PhD in 2015, during which I developed a new method for estimating the heritability of viral load in HIV.
Though the influences of many host and environmental factors on viral load are well understood, the role of the viral genome itself
in determining viral load is less clear.
I adapted a well-established method from population genetics to more accurately estimate the heritability of viral load using a phylogeny of viral sequences. This method enables analysis on incredibly large datasets, and I have investigated the viral genetic contribution to viral load in subtypes B and C in the UK HIV epidemic, using 8,483 and 1,821 sequences, respectively (provided by the UK HIV DRB). -
For my Master's (MSc) dissertation, I looked for evidence of adaptive selection in coding and
non-coding genes in Drosophila. Adaptive substitution rates in coding regions and
5' and 3' UTRs (untranslated regions) were analysed by tissue-specific, time-specific,
and immune-related gene function.
Coding regions of immune-related genes were found to have significantly higher adaptive rates than non-immune-related genes, but no difference was found in UTRs. All three regions were shown to have similar rates of adaptive evolution in most tissue-specific and time-specific genes, though UTRs had significantly higher adaptive rates than coding regions in some cases. The study provided evidence that UTRs have a faster overall adaptive rate but also more non-adaptive substitutions, and that the adaptive rate of UTRs and coding regions varies by gene function.


Graduate/Under-Graduate
-
After graduating from TCU, I worked with Dr. John Horner on the carnivorous pitcher plant
Sarracenia alata. I aided in preliminary studies on the aromatic compounds that
Sarracenia may use to attract prey, and also investigated the genetic variation
in four geographically separate populations of Sarracenia. Using AFLP analysis,
our study concluded that though long suspected to be primarily clonally reproducing,
only 14% of the genetic variation in Sarracenia alata occurred among populations,
while 86% occurred within populations, indicating that clonal spread is actually quite low
in these populations.
I presented a poster on this work at the Evolution 2010 Conference in Portland, Oregon.
"My work can be taxing and hazardous, but dull? Never."
Please find an updated list of my publications on Google Scholar.
Publications
Using nearly full-genome HIV sequence data improves phylogeny reconstruction in a simulated epidemic. Yebra G, Hodcroft EB, Ragonnet-Cronin ML,
Pillay D, Leigh Brown AJ, PANGEA_HIV, and ICONIC. Scientific Reports. 2016.
link)
Using simulated sequences, the effect of using partial and whole-genome HIV sequences and different sample
depths on reconstructing phylogenies was investigated, showing that full-genome sequences allow more reliable phylogenetic reconstruction.
Phylogenetic Tools for Generalized HIV-1 Epidemics: Findings from the PANGEA-HIV Methods Comparison. Ratmann O, Hodcroft EB, et al.,
on behalf of the PANGEA-HIV consortium. Molecular Biology and Evolution. 2016.
(link)
Two models were used to create a variety of HIV epidemic simulations. Sequences and phylogenies were publicly released and
groups were invited to try and estimate epidemic parameters such as incidence, transmissions during acute stage, and migration rate.
Identifying Transmission Clusters with Cluster Picker and HIV-TRACE. Rose R, Lamers SL, Dollar JJ, Grabowski MK, Hodcroft EB,
Ragonnet-Cronin M, Wertheim JO, Redd AD, Danielle G, and Laeyendecker O. AIDS Research and Human Retroviruses. 2016.
(link)
Two different cluster-identification approaches (Cluster Picker and HIV-TRACE) were compared on different datasets and at different genetic distances. In general,
HIV-TRACE found fewer, larger clusters, while Cluster Picker grouped sequences into more, smaller clusters.
A Direct Comparison of Two Densely Sampled HIV Epidemics: The UK and Switzerland. Ragonnet-Cronin M, Shilaih M, Gunthard HF, Hodcroft EB,
Boni J, Fearnhill E, Dunn D, Yerly S, Klimkait T, Aubert V, Yang WL, Brown AE, Lycett SJ, Kouyos R, and Leigh Brown AJ.
Scientific Reports. 2016.
(link)
The UK and Switzerland have two of the most densely sampled HIV epidemics, and collect clinical and genetic information in comprehensive national databases. In a highly collaborative effort
involving confidential and clinically sensitive data, we were able to compare the two epidemics, finding similar underlying dynamics.
Transmission of Non-B HIV Subtypes in the United Kingdom Is Increasingly Driven by Large Non-Heterosexual Transmission Clusters.
Ragonnet-Cronin M, Lycett SJ, Hodcroft EB, Hue S, Fearnhill E, Brown AE, Delpech V, Dunn D, Leigh Brown AJ, on behalf of the UK HIV DRB. Journal of Infectious Diseases. 2015.
(link)
Non-B HIV subtypes are historically associated with heterosexual transmission in the UK. However, as non-B subtypes become more prevalent, there is
evidence of crossover transmission from heterosexuals to MSM and PWID risk groups.

The Contribution of Viral Genotype to Plasma Viral Set-Point in HIV Infection. Hodcroft EB, Hadfield JD, Fearnhill E,
Phillips A, Dunn D, O'Shea S, Pillay D, Leigh Brown AJ. PLoS Pathogens. 2014.
(link)
Here we implement a new phylogenetic method to estimate the heritability of viral load in subtype B in the UK, and investigate
the change in viral load over time due to selection.
Automated Analysis of Phylogenetic Clusters. Ragonnet-Cronin M, Hodcroft EB, Hue S, Fearnhill E, Delpech V,
Leigh Brown AJ, Lycett S, on behalf of the UK HIV RDB. BMC Bioinformatics. 2013.
(link)
Two new programs are introduced to allow efficient and easy analysis of phylogenetic clusters. The
ClusterPicker allows
users to 'pick' clusters of closely related sequence by specified thresholds; the
ClusterMatcher (written by myself) allows
users to 'match' clusters containing the same sequences from two different runs, and also to investigate the attributes of
the clusters.
Presentations
3 Minute Thesis
In 2014 I had the privilege of competing in the 3 Minute Thesis competition,
presenting my work on estimating the heritability of
viral load in HIV. I advanced through the School and College levels to win both first prize and the 'people's choice' award at the
University of Edinburgh finals. I then went onto the UK Semi-Final in York, where I advanced to the UK final in Manchester alongside
five others. I also prepared a video of my presentation to compete against 17 other finalists in the world-wide
Universitas 21 competition,
where I placed 3rd.
You can view a video of my 3 Minute Thesis below!
Posters and Talks
Using DSPS-HIV Simulations and Phylogenetic Analysis to Investigate Under-sampled Hosts in the UK Heterosexual HIV Epidemic. Presented at Population Genetic Group 50 (2016) in Cambridge, UK. (Talk unavailable online)Investigating Gender Bias in HIV Transmission Clusters Using Phylogenetics and Simulation. Presented at MIDAS Network Meeting 2016 in Washington DC. (Poster unavailable online)
Using DSPS-HIV, A Highly-Customisable Agent-Based HIV Epidemic Simulator, to Investigate 'Missing Men' in Western Heterosexual HIV Epidemics. Presented at HIV Dynamics and Evolution 2016 in Woods Hole, MA. (Talk unavailable online)
Detecting Changes in Incidence Using Phylogenetic Tools: Simulation-Based Studies Within the PANGEA_HIV Initiative. Presented at CROI 2015 in Seattle, WA (abstract and poster available here).
HIV Virulence Has Not Increased in the UK Subtype B Epidemic. Presented at CROI 2014 in Boston, MA (abstract and poster available here).
I have also presented my work on heritability estimation in subtypes B and C at CROI 2012 in Seattle, WA, and CROI 2013 in Atlanta, GA. Unfortunately these posters are no longer available online. I've also spoken about this work at many conferences, including the 45th Population Genetics Group Conference in Nottingham, England, and the 19th, 20th, and 21st HIV Dynamics and Evolution Conferences in Asheville, NC; Utrecht, the Netherlands; and Tucson, AZ.
My love of programming means I'm always eager to code something, and my move to the Neher lab has truly allowed me to capitalize on this!
I'm a contributor to TreeTime and nextstrain on github,
and you can find my most recent work there.
My 'native' language is Java (which I learned in 2001), but I've recently adopted Python, and have been using R since 2009.
During my PhD and post-doc with PANGEA_HIV I also wrote a few programs (all in Java), which can be found below.
In particular, TreeCollapserCL, which collapses trees based on bootstrap support values, is the most popular program
and potentially the most useful to others.
Past Programs
*Most Popular* TreeCollapserCL 4

A new, improved version of TreeCollapseCL that can root trees and find lengths of branches
and average bootstraps of nodes, as well as collapsing nodes with bootstraps
below a user-specified threshold.
Updated: Corrects an issue with the collapsing algorithm that sometimes lead to over-collapsing. It's
highly recommended that you re-run data with TreeCollapseCL4.
PareTree 1.0.2

A basic, command-line Java program that allows users to 'pare' down their tree by either
removing unwanted sequences/leaf-nodes, removing bootstrap information, removing branch lengths - or any combination
of those three - quickly and efficiently.
Updated: Now also allows users to remove branch lengths from the tree. Also fixed a few minor bugs.
*Published* ClustMatcher

A cluster is a monophyletic group of sequences in a phylogeny that fall within specified
bootstrap support and genetic distance thresholds. In the study of infectious diseases,
especially HIV, they can represent transmission events between individuals.
Samantha Lycett's tool, ClusterPicker, is able to 'pick' clusters from a phylogeny.
The ClustMatcher tool can then be used to find clusters the contain some or all of the same
sequences between the two data sets, and outputs annotated FigTree files containing matching
clusters. This allows the change in cluster size to be compared over time.
ClustMatcher can also be used with one data set to select only clusters that contain a certain
number of sequences or have a certain attribute (clusters that contain females, for example), for
further study.
The paper detailing the ClusterMatcher and ClusterPicker software is here.
DSPS-HIV
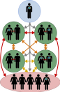
Based on the Discrete Spatial Phylo Simulator, coded by Dr. Samantha Lycett, the DSPS-HIV is a stochastic, agent-based model which has been highly modified to simulate realistic HIV epidemics. Transmission risk and disease progression rate are dependant on viral load, which is heritable, and contact networks are highly customizable. Acute, chronic, and AIDS disease stages are modelled, and treatment can be introduced at varying levels and speeds. All transmissions are tracked, so that a viral phylogeny of the epidemic is produced.
I am rarely happier than when spending an entire day programming my computer to perform automatically a task that would otherwise take me a good ten seconds to do by hand.
About Emma Hodcroft
"I’ve been travelling so long, hotels before dawn in strange cities, so long on the road that I feel the jet-speed vibration in my bones, in my body, a sense of constant motion across continents and time zones that continues long after I’m off the plane and swaying at yet another check-in desk, Hi my name is Emma."
Academic

I completed my undergraduate degree in biology at Texas Christian University (TCU), where I helped to set up and run the Purple Bike Program, a green initiative that rented free bikes to students to help reduce pollution and carbon emissions on campus. I also worked as a Java programming tutor, a job I very much enjoyed.
After graduating in December of 2008, I took a research assistant position with Dr. John Horner investigating how carnivorous Sarracenia alata pitcher plants attract their prey, as well as the genetic diversity of Sarracenia populations in the Southern US.
In the autumn of 2009, I moved from Texas to Edinburgh, Scotland, and began my master's degree at the University of Edinburgh on the Quantitative Genetics and Genome Analysis course. Though a challenging year, the course gave me an excellent introduction to the world of population and quantitative genetics.
After receiving my MSc degree with distinction in the autumn of 2010, I took a year-long research assistant position with Prof. Andrew Leigh Brown investigating virulence in HIV. Having been won-over by the wonderful world of viruses, I began my PhD with Prof. Leigh Brown in September 2011 to continue my work on HIV, and defended my thesis in May 2015.
I completed my first post-doc position with the PANGEA_HIV initiative, continuing in Andrew Leigh Brown's lab, where I devloped a realistic, stochastic agent-based model to simulate HIV epidemics in sub-Saharan Africa.
I am currently a post-doc working on nextstrain with Richard Neher - you can find out more under 'Home' or 'Research'.
Personal

Born in Norway, and raised spending half the year in Scotland with my father and half the year in Texas with my mother, I'm a strange mix of two countries more similar than one might expect!
My half-and-half upbringing has given me a unique perspective on life, as well as an interesting vocabulary and an amusing accent. A fan of both kilts and cowboy boots, I feel equally at home in both places.
I'm lucky enough to have had the opportunity to travel around North and South America, Europe, and even venture a little into Asia. My bi-annual migrations between Texas and Scotland all my life mean I'm quite at home in airports and on planes, and am no stranger to travel at all.
As well as my love of biology, evolution and programming, I'm a proud feminist, and very much enjoy a good debate on any controversial topic. I love reading a wide variety of books, from popular fiction and 'pop-sci' to non-fiction and classics. Being a third-generation computer geek, I enjoy all things tech-y and have had a deep love of programming since I was 15. I can often be found gaming - usually Zelda, Overwatch, and Vermintide!
I played violin regularly in various orchestras from age 10 to 21 and still enjoy it, though I don't play as much as I'd like to. Finally, I have a fondness for the colour purple, cephalopods, airplanes, potatoes, and cats.

I can be contacted at the address below:
The prevalence of spam-bots keeps me from posting my email address, but you can contact me via the feedback form.
“Science may never come up with a better office communication system than the coffee break.”